scRNAseq_complete_04_Annotation_v7
retogerber
2024-01-22
Last updated: 2024-01-22
Checks: 6 1
Knit directory: synovialscrnaseq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210105) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 58eeb06. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: '/
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .empty/
Ignored: analysis/.Rhistory
Ignored: analysis/iSEE_interactive_document.html
Ignored: code/test_files/
Ignored: data/Culemann/
Ignored: data/E-MTAB-8322/
Ignored: data/Synovial scRNA-seq samples - Sheet1.csv
Ignored: data/Zhang_top20_singlecell_cluster_markers_fromGithub.csv
Ignored: data/findMarkers_results.rds
Ignored: data/findMarkers_results_v2.rds
Ignored: data/info/
Ignored: data/syn_sce_tidy_filtered.rds
Ignored: data/syn_sce_tidy_hvg.rds
Ignored: data/syn_sce_tidy_hvg_cms.rds
Ignored: docs/
Ignored: output/Figures_Paper/
Ignored: output/Sample_summaries_RA_comparisons.rds
Ignored: output/Sample_summaries_direct_dissociation.rds
Ignored: output/Sample_summaries_exvivo_treatment.rds
Ignored: output/Suppl_Figure_4d.rds
Ignored: output/barcodes.txt
Ignored: output/barcodes_filtered.txt
Ignored: output/column_metadata_filtered.txt
Ignored: output/combined_v7_SingleR_markers.rds
Ignored: output/combined_v7_SingleR_predictions.rds
Ignored: output/combined_v7_SingleR_predictions_lclc.rds
Ignored: output/combined_v7_SingleR_predictions_reclc.rds
Ignored: output/combined_v7_SingleR_predictions_recrec.rds
Ignored: output/combined_v7_SingleR_trained.rds
Ignored: output/combined_v7_sce.rds
Ignored: output/combined_v7_sce_filtered.rds
Ignored: output/combined_v7_sce_hvg.rds
Ignored: output/combined_v7_sce_hvg_cms.rds
Ignored: output/combined_v7_sce_hvg_cms_annotated.rds
Ignored: output/combined_v7_sce_tmp_hvg_cms.rds
Ignored: output/combined_v7_upsetplot_genelists.rds
Ignored: output/count_matrix_filtered.mtx
Ignored: output/count_matrix_unfiltered.mtx
Ignored: output/emptyDrops_result_v4.rds
Ignored: output/emptyDrops_result_v4_tmp.rds
Ignored: output/emptyDrops_result_v4tmptmp.rds
Ignored: output/findMarkers_results_v6.rds
Ignored: output/findMarkers_results_v6_ec.rds
Ignored: output/findMarkers_results_v6_main.rds
Ignored: output/findMarkers_results_v6_mp.rds
Ignored: output/findMarkers_results_v6_sf.rds
Ignored: output/findMarkers_results_v6_tc.rds
Ignored: output/findMarkers_results_v7_ec.rds
Ignored: output/findMarkers_results_v7_main.rds
Ignored: output/findMarkers_results_v7_mp.rds
Ignored: output/findMarkers_results_v7_sf.rds
Ignored: output/findMarkers_results_v7_tc.rds
Ignored: output/genes.txt
Ignored: output/genes_filtered.txt
Ignored: output/goana_results_v6_ec.rds
Ignored: output/goana_results_v6_mp.rds
Ignored: output/preprocessing_number_of_cells.rds
Ignored: output/syn_v4_sce_emptyDrops_invivo.rds
Ignored: output/syn_v4_swappedDrops_24300_after.rds
Ignored: output/syn_v4_swappedDrops_24300_before.rds
Ignored: output/syn_v4_swappedDrops_24793_after.rds
Ignored: output/syn_v4_swappedDrops_24793_before.rds
Ignored: output/syn_v6_cluster_cellid_match_invivo.rds
Ignored: output/syn_v6_clustering_lookup_invivo.rds
Ignored: output/syn_v6_clustering_lookup_multiple_invivo.rds
Ignored: output/syn_v6_sce.rds
Ignored: output/syn_v6_sce_Figure8.rds
Ignored: output/syn_v6_sce_Figure8_dic_ls.rds
Ignored: output/syn_v6_sce_ec_invivo.rds
Ignored: output/syn_v6_sce_filtered_invivo.rds
Ignored: output/syn_v6_sce_hdf5/
Ignored: output/syn_v6_sce_hvg_cms_doublet_invivo.rds
Ignored: output/syn_v6_sce_hvg_cms_doublet_subcluster_invivo.rds
Ignored: output/syn_v6_sce_hvg_invivo.rds
Ignored: output/syn_v6_sce_hvg_marker_genes.rds
Ignored: output/syn_v6_sce_mp_invivo.rds
Ignored: output/syn_v6_sce_sf_invivo.rds
Ignored: output/syn_v6_sce_tc_invivo.rds
Ignored: output/syn_v6_sfig1.rds
Ignored: output/syn_v6_vst_out_invivo.rds
Ignored: output/syn_v7_cluster_cellid_match_invivo.rds
Ignored: output/syn_v7_clustering_lookup_invivo.rds
Ignored: output/syn_v7_clustering_lookup_multiple_invivo.rds
Ignored: output/syn_v7_sce.rds
Ignored: output/syn_v7_sce_Figure8.rds
Ignored: output/syn_v7_sce_Figure8_dic_ls.rds
Ignored: output/syn_v7_sce_ec_invivo.rds
Ignored: output/syn_v7_sce_ec_invivo_trajectory.rds
Ignored: output/syn_v7_sce_ec_invivo_trajectory2.rds
Ignored: output/syn_v7_sce_ec_invivo_trajectory2_ATres.rds
Ignored: output/syn_v7_sce_ec_invivo_trajectory_icMat.rds
Ignored: output/syn_v7_sce_filtered_invivo.rds
Ignored: output/syn_v7_sce_hdf5/
Ignored: output/syn_v7_sce_hvg_cms_doublet_invivo.rds
Ignored: output/syn_v7_sce_hvg_cms_doublet_subcluster_invivo.rds
Ignored: output/syn_v7_sce_hvg_cms_doublet_subcluster_invivo_cleaned.rds
Ignored: output/syn_v7_sce_hvg_invivo.rds
Ignored: output/syn_v7_sce_mp_invivo.rds
Ignored: output/syn_v7_sce_sf_invivo.rds
Ignored: output/syn_v7_sce_tc_invivo.rds
Ignored: output/syn_v7_sfig1.rds
Ignored: output/syn_v7_vst_out_invivo.rds
Untracked files:
Untracked: analysis/clean_and_save_sce.R
Untracked: analysis/description_integration_wei_stephenson
Untracked: analysis/scRNAseq_complete_01_preprocessing_comparison.Rmd
Untracked: analysis/scRNAseq_complete_05_ec_trajectory_analysis.Rmd
Untracked: analysis/scRNAseq_complete_05_ec_trajectory_analysis_2.Rmd
Untracked: analysis/scRNAseq_complete_05_ec_trajectory_analysis_3.Rmd
Untracked: code/rebuild_ezRun.R
Untracked: nonhosted_public/
Untracked: singRstudio.sh.bak
Unstaged changes:
Modified: analysis/scRNAseq_combined_06_Figures.Rmd
Modified: analysis/scRNAseq_complete_04-2_celltype_markers.Rmd
Modified: analysis/scRNAseq_complete_04-2_celltype_markers_subcelltypes.Rmd
Modified: analysis/scRNAseq_complete_04_Annotation_v7.Rmd
Modified: analysis/scRNAseq_complete_Figures.Rmd
Modified: analysis/write_tsv.Rmd
Modified: code/create_hdf5.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/scRNAseq_complete_04_Annotation_v7.Rmd) and HTML (public/scRNAseq_complete_04_Annotation_v7.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 58eeb06 | Reto Gerber | 2023-05-30 | add new version |
| html | 58eeb06 | Reto Gerber | 2023-05-30 | add new version |
| Rmd | 4575ba6 | Reto Gerber | 2022-12-21 | Update analyis |
Set up
suppressPackageStartupMessages({
library(magrittr)
library(SingleCellExperiment)
})
n_workers <- 10
RhpcBLASctl::blas_set_num_threads(n_workers)
analysis_version <- 7
remove_low_quality_samples <- TRUE
here::here()[1] "/home/retger/Synovial/synovialscrnaseq"set.seed(100)
clusters_lookup <- list()sce_main <- readRDS(file =here::here("output",paste0("syn_v",analysis_version,"_sce_hvg_cms_doublet_subcluster_invivo.rds")))sce_main$Diagnosis_main[sce_main$Sample%in%c("Syn_Bio_078","Syn_Bio_091","Syn_Bio_099")] <- "Undiff. Arthritis"Loading required package: tidySingleCellExperiment
Attaching package: 'tidySingleCellExperiment'The following object is masked from 'package:IRanges':
sliceThe following object is masked from 'package:S4Vectors':
renameThe following object is masked from 'package:matrixStats':
countThe following object is masked from 'package:magrittr':
extractThe following object is masked from 'package:stats':
filterAnnotation main celltypes
sce_main$kgraph_clusters_final <- sce_main$kgraph_clusters
scater::plotReducedDim(sce_main,"UMAP_corrected", colour_by = "kgraph_clusters_final",text_by="kgraph_clusters_final")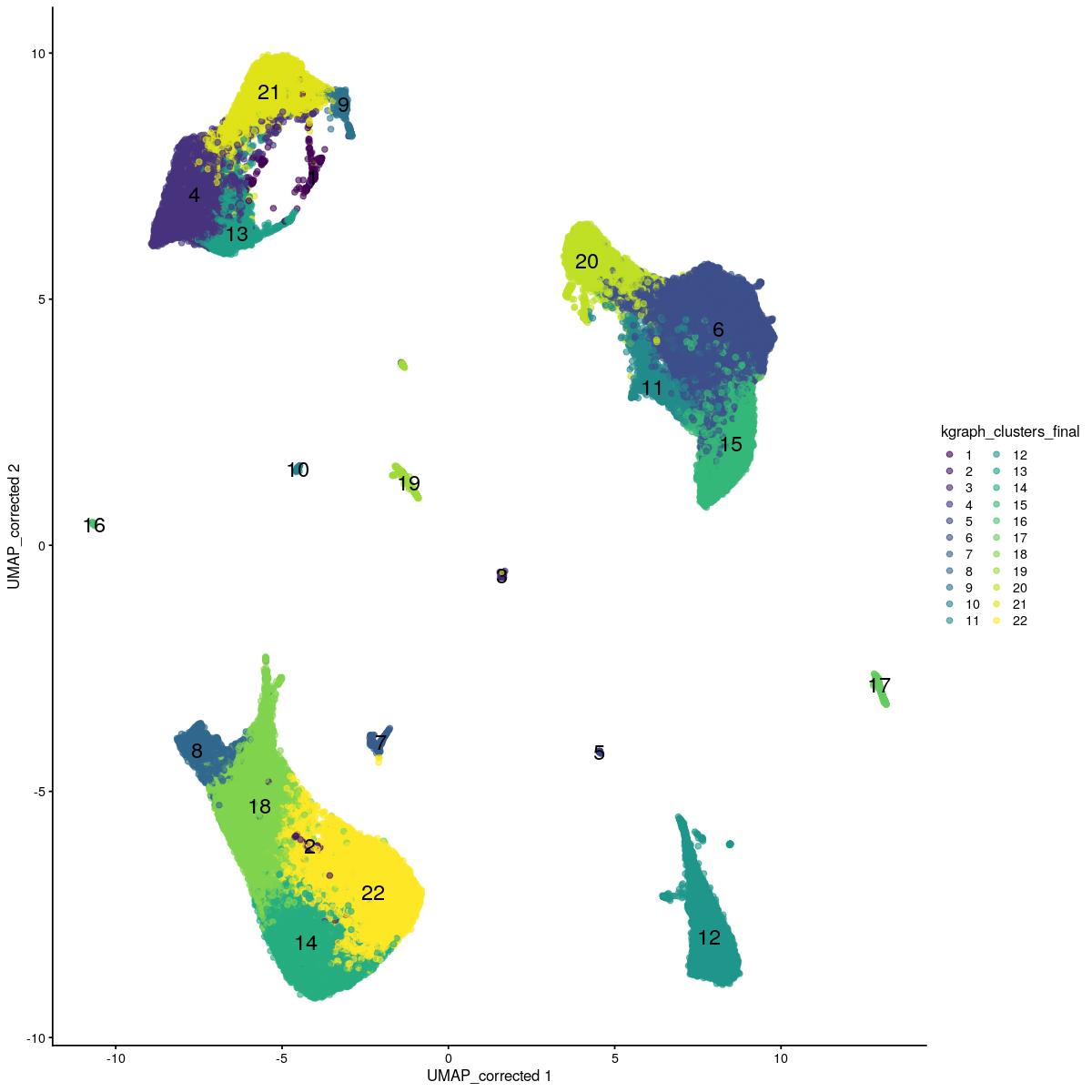
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
scater::plotReducedDim(sce_main[,sce_main$kgraph_clusters_final==19],"UMAP_corrected", colour_by = "kgraph_clusters_final",text_by="kgraph_clusters_final")Warning: Removed 21 rows containing missing values (geom_text).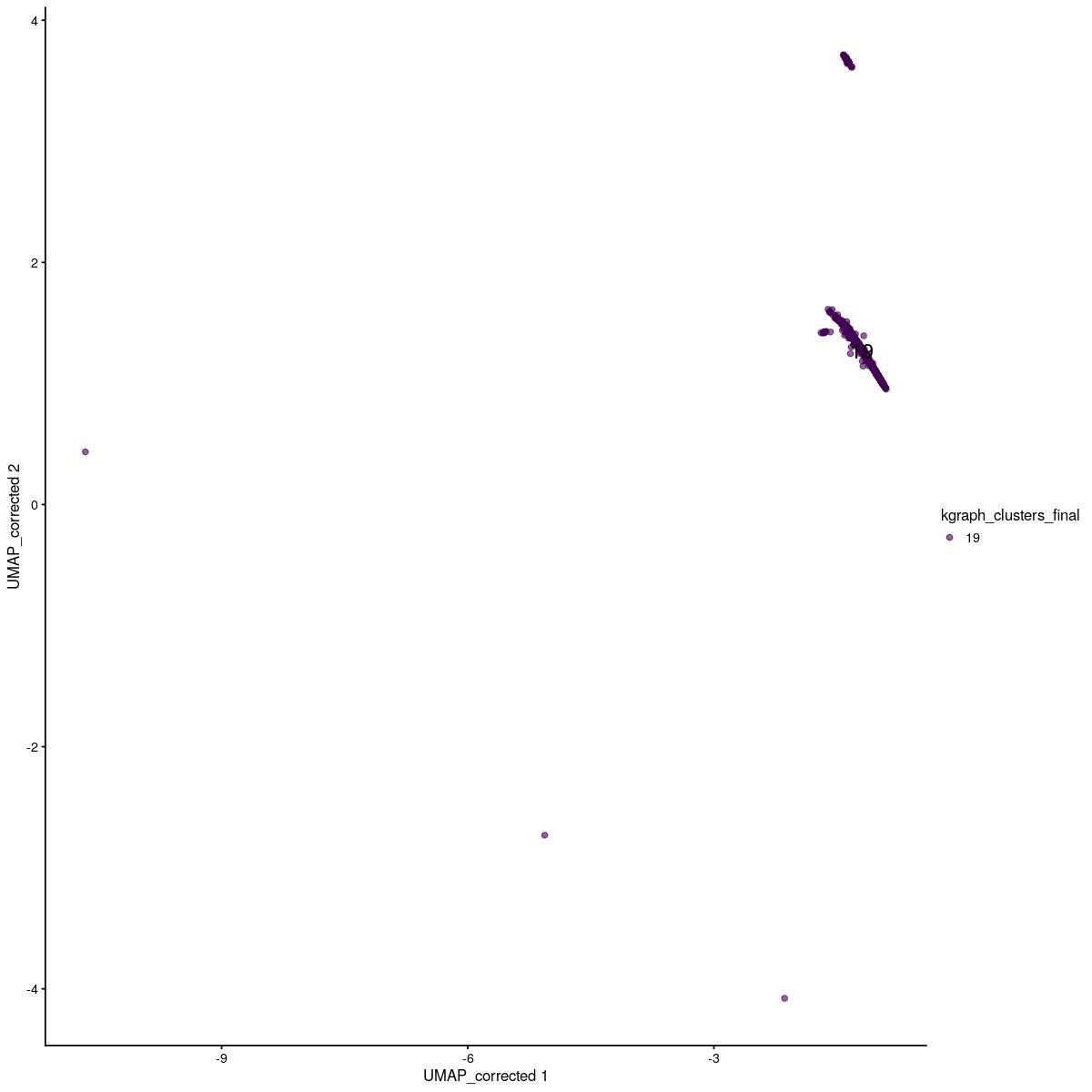
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
tmp <- sce_main[,sce_main$kgraph_clusters_final==19]
set.seed(123)
kout <- kmeans(reducedDim(tmp,"corrected"),2)
tmp$kout <- kout$cluster
scater::plotReducedDim(tmp,"UMAP_corrected", colour_by = "kout")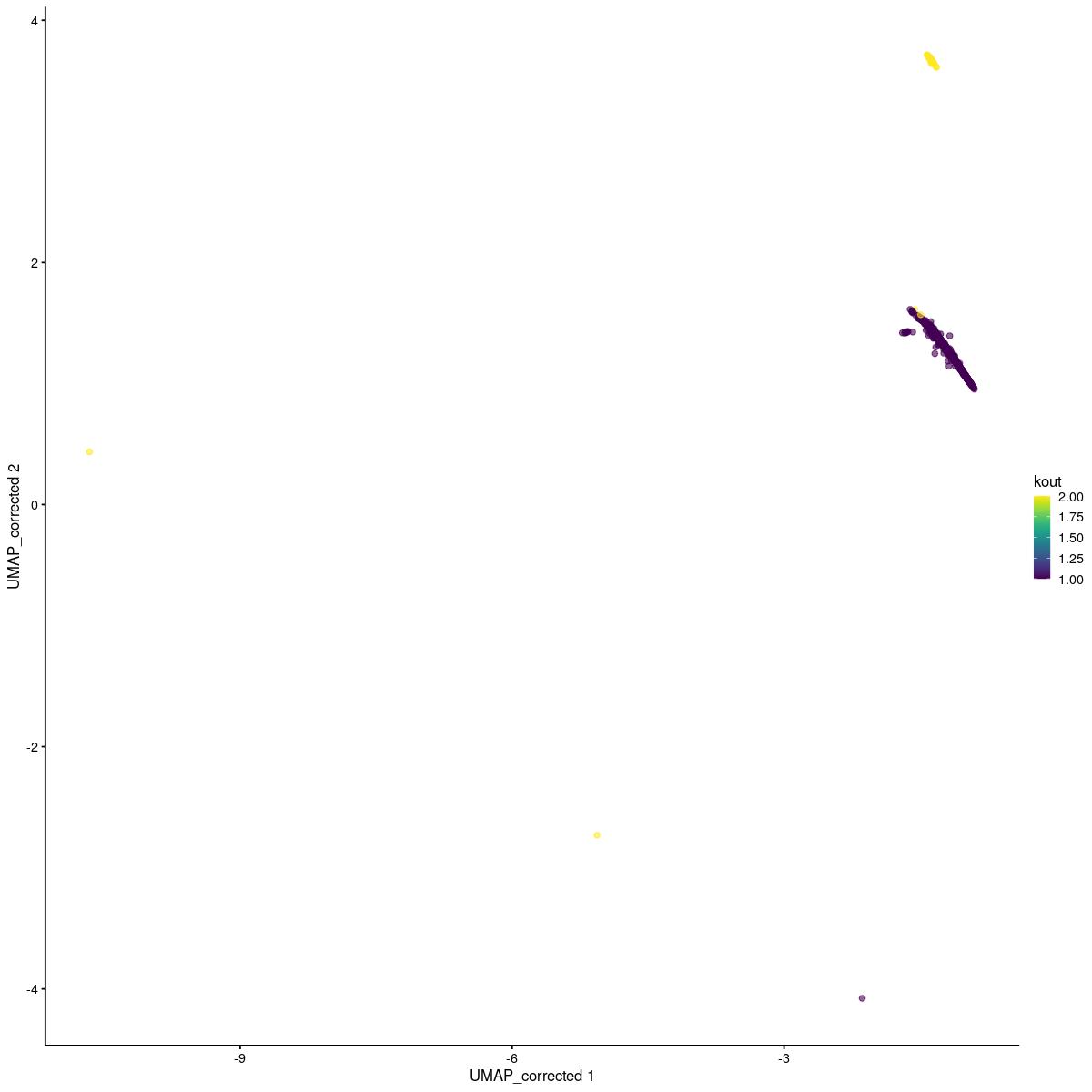
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
levels(sce_main$kgraph_clusters_final) <- c(levels(sce_main$kgraph_clusters_final),"23")
sce_main$kgraph_clusters_final[sce_main$kgraph_clusters_final == 19][tmp$kout==2] <- 23sce_main$main_celltype <- as.integer(sce_main$kgraph_clusters_final)
sce_main$main_celltype[sce_main$main_celltype %in% c(1,4,9,13,21)] <- "T cells/NK cells"
sce_main$main_celltype[sce_main$main_celltype %in% c(5,12)] <- "Endothelial cells"
sce_main$main_celltype[sce_main$main_celltype %in% c(6,11,15,20)] <- "Fibroblasts"
sce_main$main_celltype[sce_main$main_celltype %in% c(2,8,14,18,22)] <- "Macrophages"
sce_main$main_celltype[sce_main$main_celltype %in% c(7)] <- "Macrophages"
sce_main$main_celltype[sce_main$main_celltype %in% c(19)] <- "B cells"
sce_main$main_celltype[sce_main$main_celltype %in% c(17)] <- "Pericytes/Mural cells"
sce_main$main_celltype[sce_main$main_celltype %in% c(3)] <- "Neutrophils"
sce_main$main_celltype[sce_main$main_celltype %in% c(10)] <- "Mast cells"
sce_main$main_celltype[sce_main$main_celltype %in% c(16)] <- "Plasmacytoid DCs"
sce_main$main_celltype[sce_main$main_celltype %in% c(23)] <- "Plasmablasts"
scater::plotReducedDim(sce_main,"UMAP_corrected", colour_by = "main_celltype",text_by="main_celltype")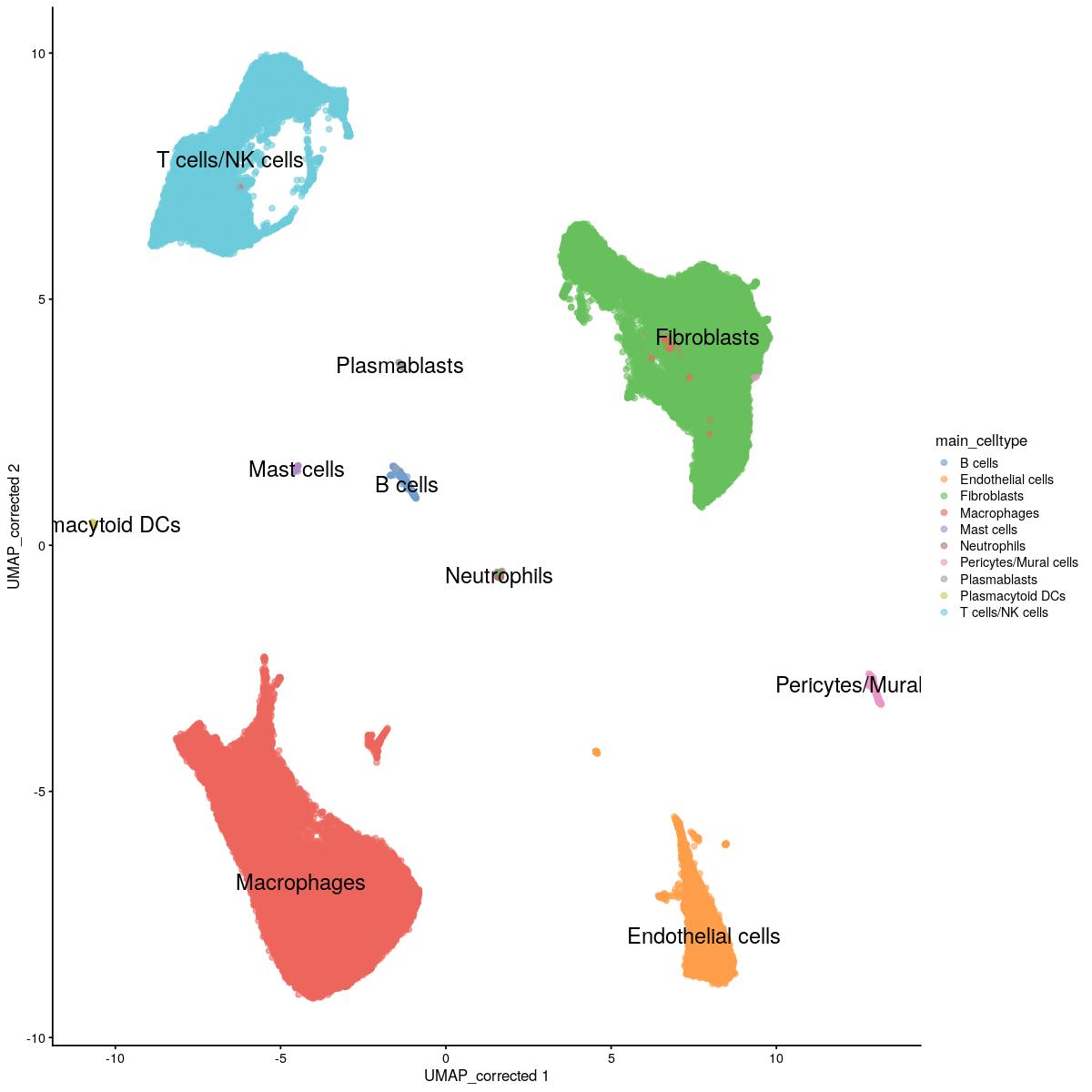
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
# sce_main$main_celltype[sce_main$main_celltype %in% c(12)] <- "Plasmablasts"
# sce_main$main_celltype[sce_main$main_celltype %in% c(10)] <- "Plasmacytoid DCs"Annotation SF
celltype_name_pre <- "sf"tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
sce_sub <- readRDS(file = here::here("output",tmpfilename))sce_sub$sf_clusters_final <- as.integer(sce_sub$sf_clusters_k30)
sce_sub$sf_celltype <- as.integer(sce_sub$sf_clusters_final)
sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(6)] <- "GGT5high CXCL12 high FGF7+"
sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(4)] <- "SERPINE1+ COL5A3+ LOXL2high"
sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(2)] <- "CADM1high ACAN+ DKK3+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(4)] <- ""
sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(3)] <- "PRG4+ CD55+ TWISTNB+ lining SF"
sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(5)] <- "TNXBhigh IGFBP6+ FGFBP2+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(1)] <- "NOTCH3+ GGT5low"
sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(1)] <- "MMP13+"
sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(7)] <- "HLA-DRAhigh CD74+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(1)] <- "GGT5high CXCL12high FGF7+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(2)] <- "SERPINE1 + COL5A3+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(3)] <- "CADM1high ACAN+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(4)] <- "SCD4+ SAA1+ SAA2+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(5)] <- "CLIC5+ HBEGF+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(6)] <- "TNXBhigh GFBP6+ FGFBP2+"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(7)] <- "NOTCH3+ GGT5low"
# sce_sub$sf_celltype[sce_sub$sf_celltype %in% c(8)] <- "HLA-DRAhigh"
clusters_lookup[[celltype_name_pre]] <- data.frame(cell_id = colnames(sce_sub),
cluster = sce_sub[["sf_celltype"]])tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
saveRDS(sce_sub, file = here::here("output",tmpfilename))Annotation MP
celltype_name_pre <- "mp"tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
sce_sub <- readRDS(file = here::here("output",tmpfilename))sce_sub$mp_clusters_final <- as.integer(sce_sub$mp_clusters_k10)
scater::plotReducedDim(sce_sub,"UMAP_corrected", colour_by = "mp_clusters_final",text_by="mp_clusters_final")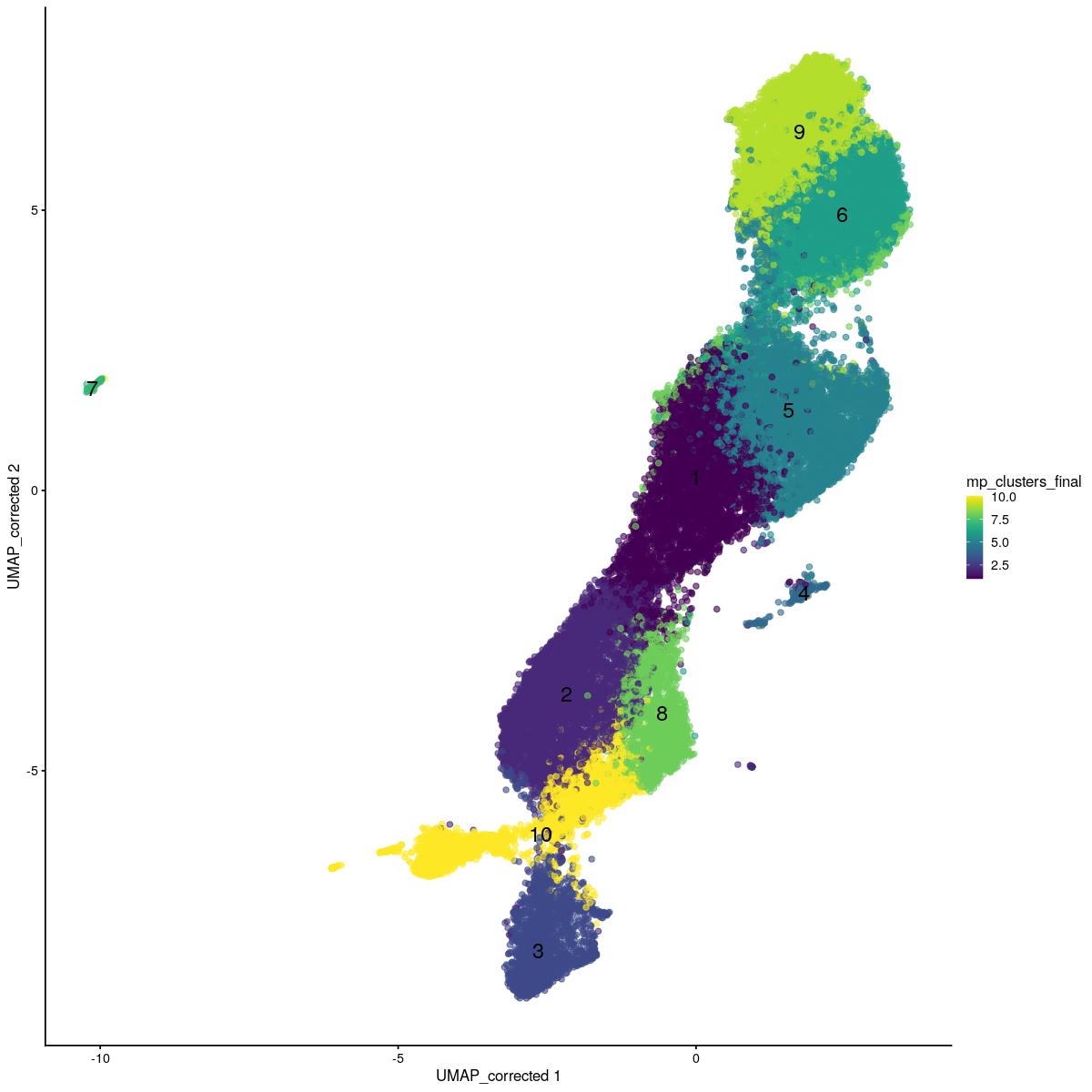
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
tmp <- sce_sub[,sce_sub$mp_clusters_final == 10]
set.seed(123)
kout <- kmeans(reducedDim(tmp,"corrected"),3)
tmp$kout <- kout$cluster
scater::plotReducedDim(tmp,"UMAP_corrected", colour_by = "kout")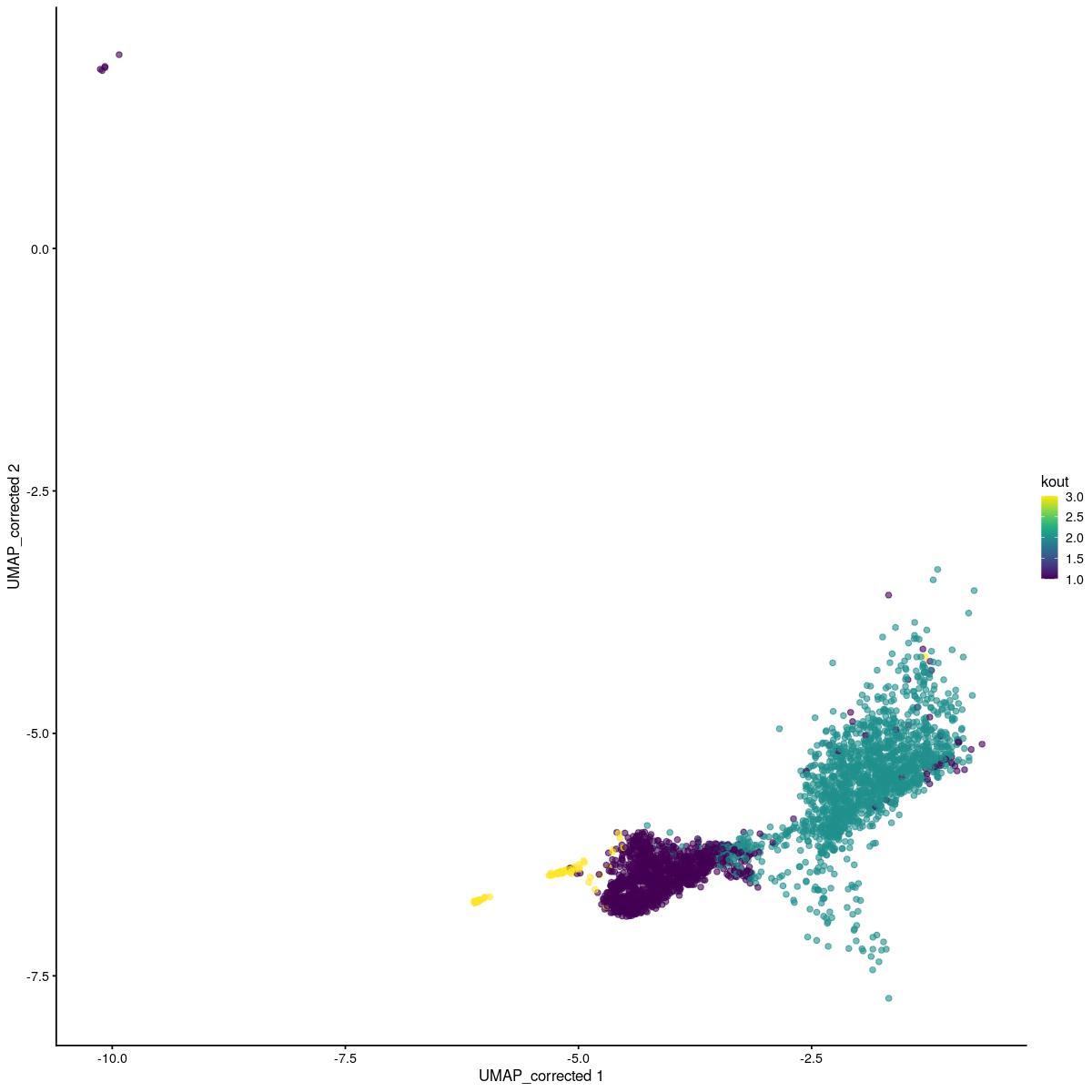
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
sce_sub$mp_clusters_final[sce_sub$mp_clusters_final == 10][tmp$kout==1] <- 11
sce_sub$mp_clusters_final[sce_sub$mp_clusters_final == 10][tmp$kout[tmp$kout!=1]==3] <- 12
scater::plotReducedDim(sce_sub,"UMAP_corrected", colour_by = "mp_clusters_final",text_by="mp_clusters_final")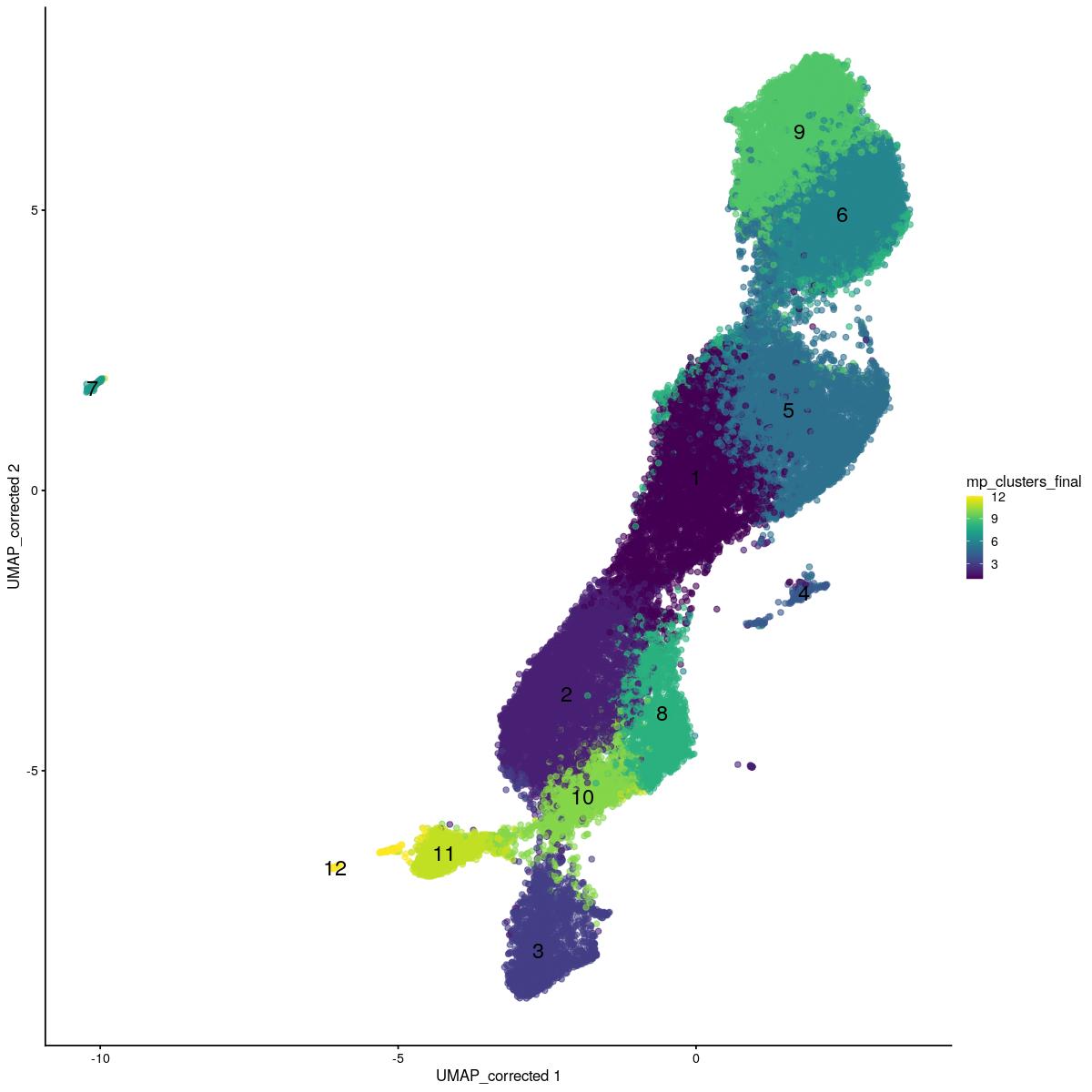
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
sce_sub$mp_celltype <- as.integer(sce_sub$mp_clusters_final)
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(1)] <- "COLEC12med TIMD4+ SPP1neg & COLEC12med TIMD4neg SPP1+"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(1)] <- "FOLR2+ MERTK+ TIMD4+ & FOLR2low MERTKlow SPP1+ subsets"
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(2)] <- "SPP1+ CD48+"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(2)] <- "CD48low SPP1+"
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(3)] <- "S100A12+ PLAC8+ CD48high"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(3)] <- "CD48high S100A12+ IL1B+"
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(4)] <- "COLEC12high TIMD4+ TOP2A+"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(4)] <- "FOLR2low MERTKlow TOP2A+ CENPF+ proliferating"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(5)] <- "FOLR2high MERTK+ SELENOPhigh COLEC12high TIMD4+"
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(6)] <- "COLEC12med CD48low"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(6)] <- "FOLR2high MERTK+ SELENOPhigh CD48med"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(7)] <- "CLEC9A+ CADM1+ CLNK+"
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(8)] <- "COLEC12neg CD48+"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(8)] <- "C1QA/B/C+ FOLR2low CCR2+ CD48+ CLEC10A"
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(9)] <- "COLEC12high CD209+ LYVE1+"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(9)] <- "FOLR2high MERTK+ SELENOPhigh COLEC12high LYVE1+ CD209+ SLC40A1+"
# sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(10)] <- "CLEC10A+ CD48low"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(10)] <- "CD48+ CLEC10A+"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(11)] <- "CD1C+ CLEC10A+"
sce_sub$mp_celltype[sce_sub$mp_celltype %in% c(12)] <- "IDO1+ LAMP3+"
clusters_lookup[[celltype_name_pre]] <- data.frame(cell_id = colnames(sce_sub),
cluster = sce_sub[["mp_celltype"]])dendritic_clusters <- c(7,11,12)
is_dendritic <- sce_sub[["mp_clusters_final"]] %in% dendritic_clusters
dendritic_cluster_loopup <- data.frame(cell_id = colnames(sce_sub)[is_dendritic], cluster = sce_sub[["mp_celltype"]][is_dendritic])
celllabelmatch <- match(dendritic_cluster_loopup$cell_id,
colnames(sce_main))
print(table(is.na(celllabelmatch)))
FALSE
1693 celllabelmatch <- celllabelmatch[!is.na(celllabelmatch)]
colData(sce_main)$main_celltype[celllabelmatch] <- "Dendritic cells"scater::plotReducedDim(sce_main,"UMAP_corrected", colour_by = "main_celltype",text_by="main_celltype")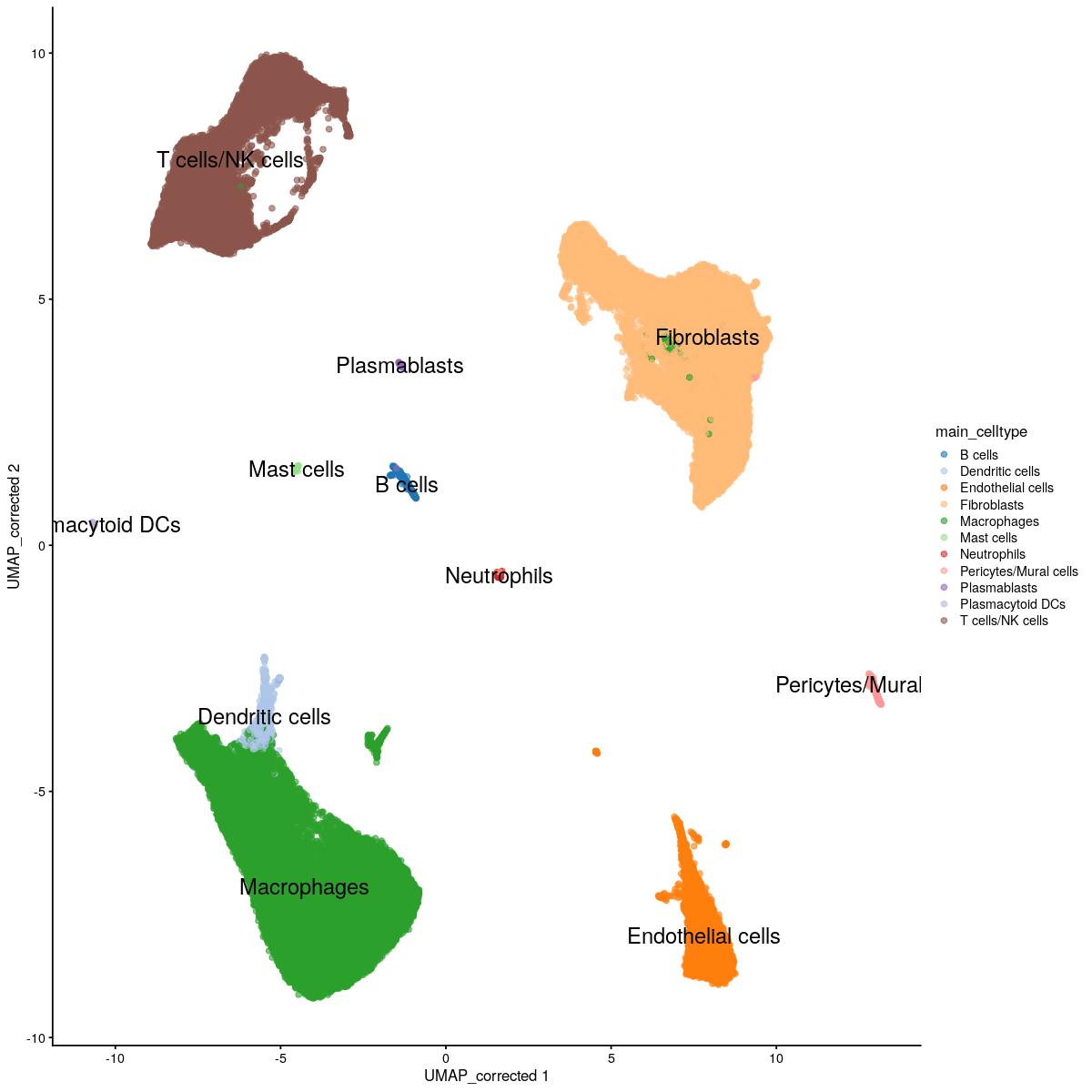
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
saveRDS(sce_sub, file = here::here("output",tmpfilename))Annotation EC
celltype_name_pre <- "ec"tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
sce_sub <- readRDS(file = here::here("output",tmpfilename))sce_sub$ec_clusters_final <- as.integer(sce_sub$ec_clusters_k20) - 1
sce_sub$ec_celltype <- as.integer(sce_sub$ec_clusters_final)
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(0)] <- NA
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(1)] <- "ACKRhigh IL1R1low CLU+ venous"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(1)] <- "ACKRhigh IL1R1+ CLU+ VCAN+ venous"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(2)] <- "GJA4+ CLDN5+ arterial"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(3)] <- "ACKRmed IL1R1- CLU- SPARChigh SELE+ transitional"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(3)] <- "ACKRmed CLU- SPARChigh"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(4)] <- "LYVE1+ PROX1+ CCL21+ lymphatic"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(5)] <- "ACKRhigh IL1R1high CLU+ SELEhigh TNFAIP3+ IL6+ CCL2high venous"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(5)] <- "ACKRhigh IL1R1+ CLU+ SELEhigh TNFAIP3+ venous"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(6)] <- "KDR+ SPP1+ SPARChigh capillary"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(7)] <- "TOP2A+ CENPF+ proliferating"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(8)] <- "ACKRhigh IL1R1med CLU+ venous"
sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(8)] <- "ACKRhigh IL1R1+ CLU+ SELE+ venous"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(1)] <- "PROX1+ LYVE1+ CCL21+ lymphatic ECs"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(2)] <- "TOP2A+ CENPF+ MKI67+ ECs"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(3)] <- "GJA4+ CLDN5+ arterial ECs"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(4)] <- "SPP1+ KDR+ capillary ECs"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(5)] <- "ACKR1high VWFhigh SELE+ IL6+ venous ECs"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(6)] <- "ACKR1high VWFhigh SELE+ IL6- venous ECs"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(7)] <- "ACKR1med KDRlow SPARChigh ECs"
# sce_sub$ec_celltype[sce_sub$ec_celltype %in% c(8)] <- "ACKR1high VWFhigh SELE+ CCL2+ TNFAIP3+ venous ECs"
clusters_lookup[[celltype_name_pre]] <- data.frame(cell_id = colnames(sce_sub),
cluster = sce_sub[["ec_celltype"]])
dim(sce_sub)[1] 17057 9378sce_sub <- sce_sub[,!is.na(sce_sub$ec_celltype)]
dim(sce_sub)[1] 17057 9378tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
saveRDS(sce_sub, file = here::here("output",tmpfilename))Annotation TC
celltype_name_pre <- "tc"tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
sce_sub <- readRDS(file = here::here("output",tmpfilename))sce_sub$tc_clusters_final <- as.integer(sce_sub$tc_clusters_k20)
# overall cluster
scater::plotReducedDim(sce_sub,"UMAP_corrected", colour_by = "tc_clusters_final")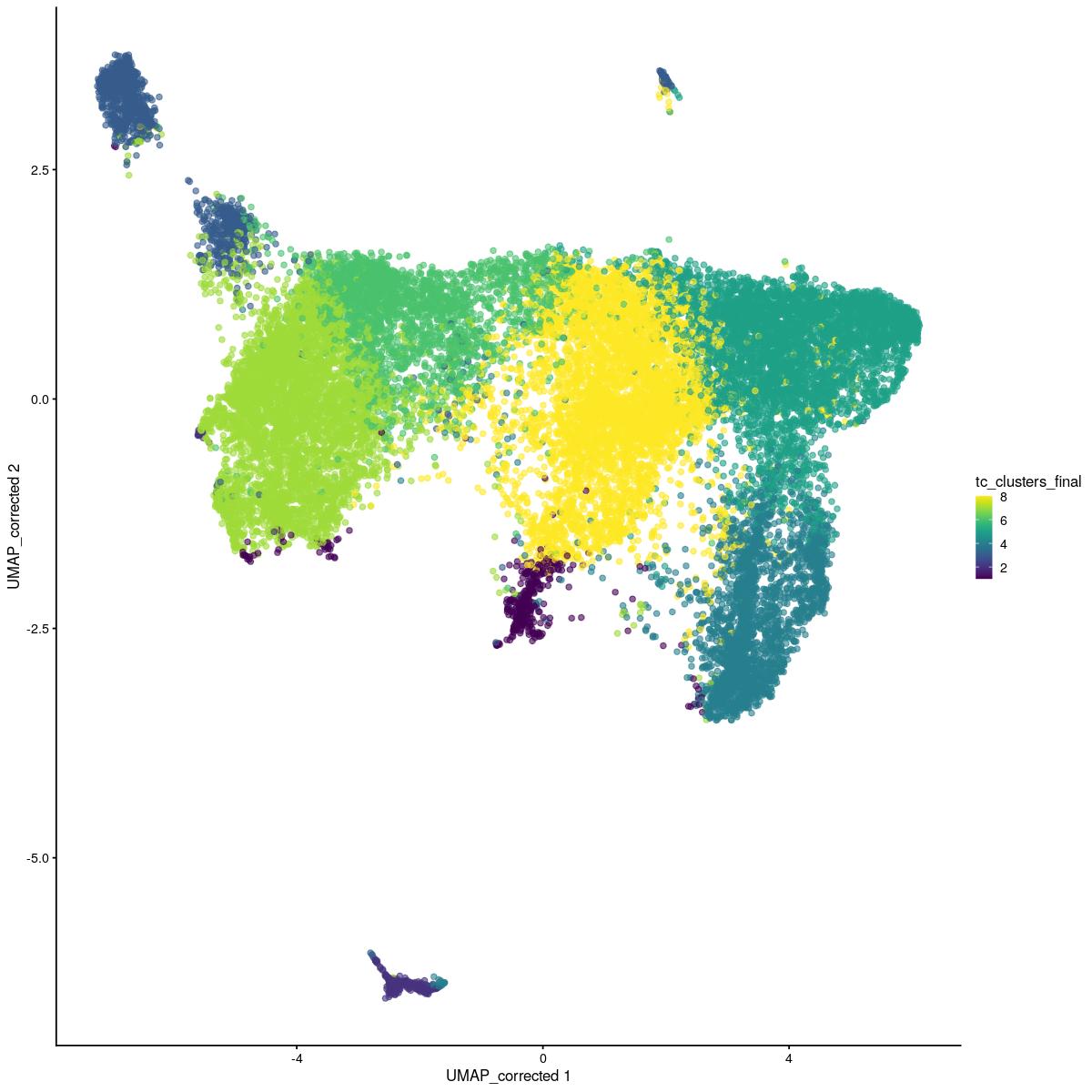
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
# merge cluster 1 and 8
sce_sub$tc_clusters_final[sce_sub$tc_clusters_final==8] <- 1
# add cluster number 10, in top right of umap
is_10 <- reducedDim(sce_sub,"UMAP_corrected")[,1] > 0 & reducedDim(sce_sub,"UMAP_corrected")[,2] > 2.5
sce_sub$is_10 <- is_10
scater::plotReducedDim(sce_sub,"UMAP_corrected", colour_by = "is_10")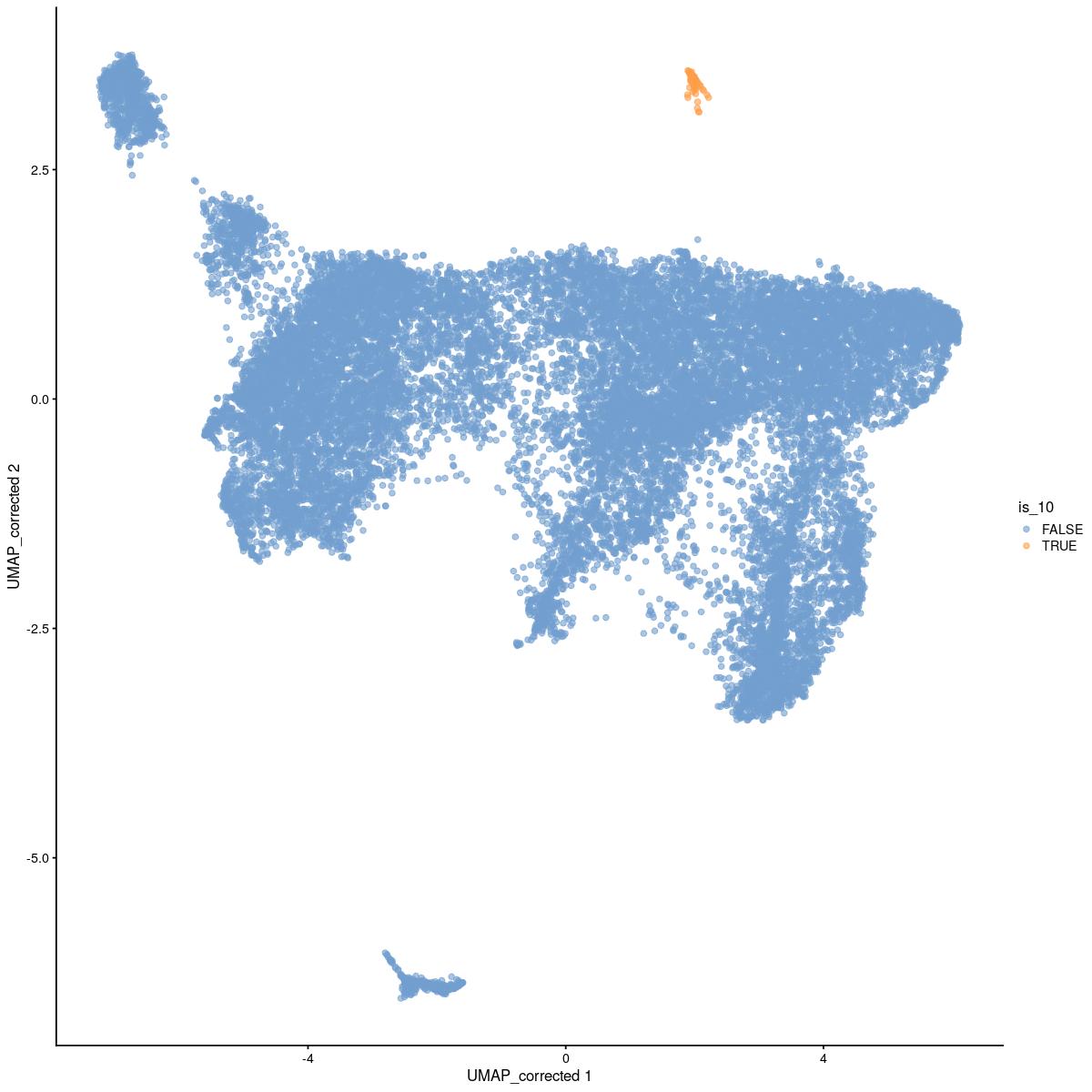
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
sce_sub$tc_clusters_final[sce_sub$is_10] <- 9
# split cluster number 3 into two with kmeans, new cluster is number 9
tmp <- sce_sub[,sce_sub$tc_clusters_final == 3]
set.seed(123)
kout <- kmeans(reducedDim(tmp,"corrected"),2)
tmp$kout <- kout$cluster
scater::plotReducedDim(tmp,"UMAP_corrected", colour_by = "kout")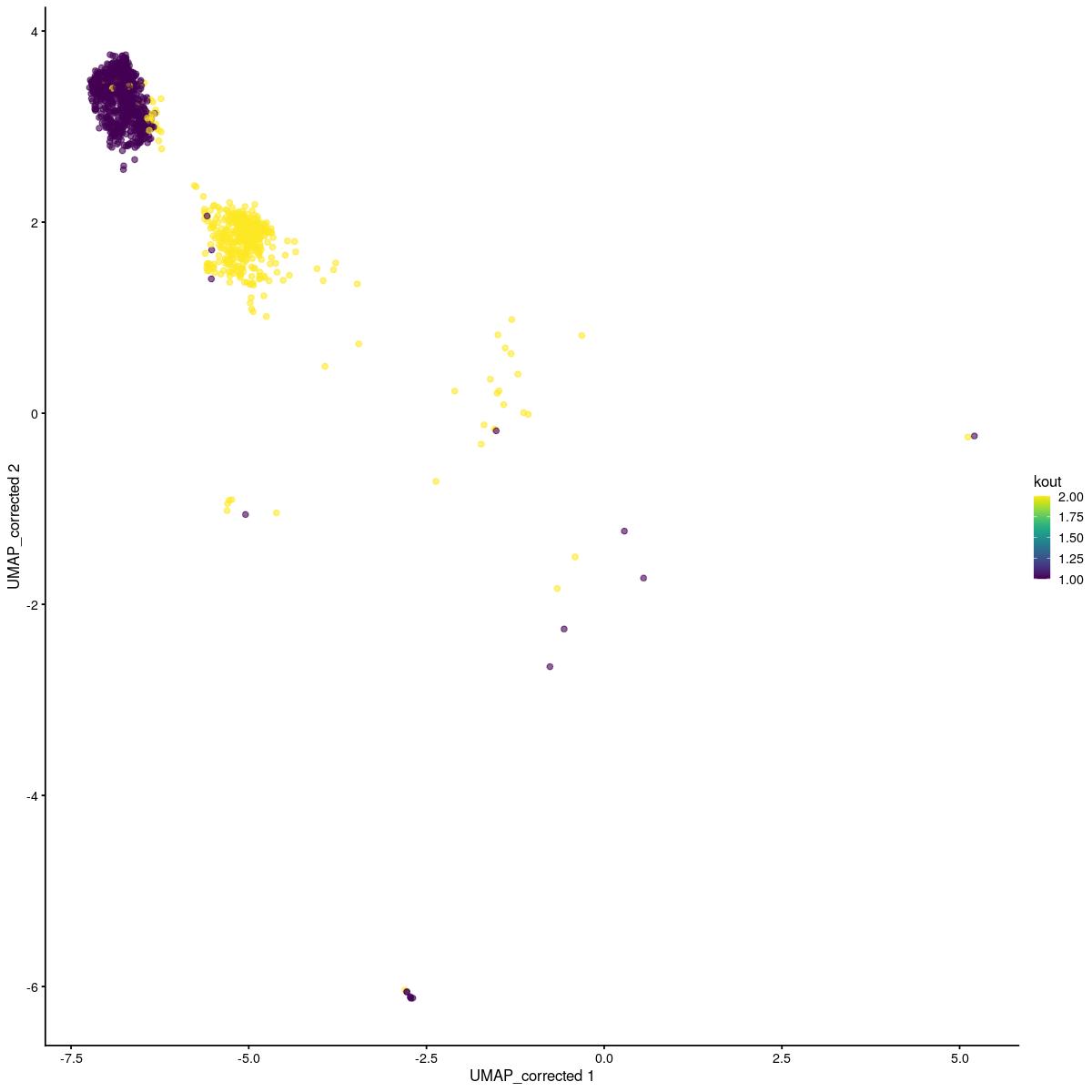
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
sce_sub$tc_clusters_final[sce_sub$tc_clusters_final == 3][tmp$kout==2] <- 8
# resulting clusters
scater::plotReducedDim(sce_sub,"UMAP_corrected", colour_by = "tc_clusters_final")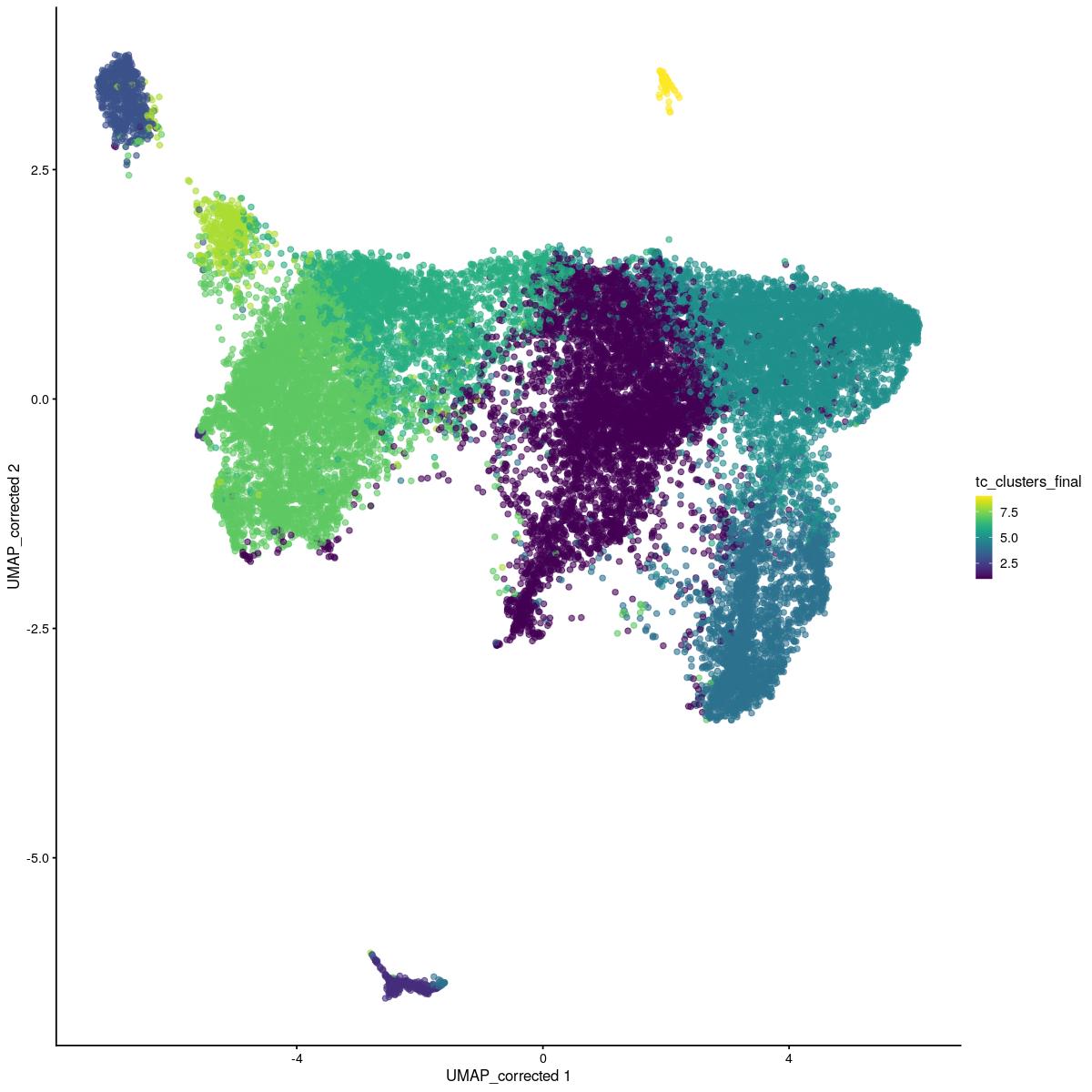
| Version | Author | Date |
|---|---|---|
| 58eeb06 | Reto Gerber | 2023-05-30 |
sce_sub$tc_celltype <- as.integer(sce_sub$tc_clusters_final)
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(1)] <- "CCR7+ CCL5+ LEF1low SELLlow"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(1)] <- "CCR7med LEF1low SELLlow"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(2)] <- "TOP2A+ CENPF+ proliferating T & NK cells"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(2)] <- "TOP2A+ CENPF+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(3)] <- "CD3- NKG7+ GNLY+ NK cells"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(3)] <- "CD3- NKG7+ GNLY+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(4)] <- "CCR7- TIGIT+ CTLA4+"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(4)] <- "TIGIT+ CTLA4+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(5)] <- "CCR7+ CCL5- LEF1+ SELL+"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(5)] <- "CCR7high LEF1+ SELL+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(6)] <- "GNLY- GZMK+ GZMH- GZMBlow"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(6)] <- "GZMB- GZMH- GZMK+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(7)] <- "GNLY- GZMK+ GZMH+ GZMB+"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(7)] <- "GZMB+ GZMH+ GZMK+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(8)] <- "NKG7+ GNLY+ GZMK- GZMB+"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(8)] <- "GZMB+ GZMH+ GZMK- GNLY+"
sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(9)] <- "CD3- NKG7- KLRB1+ IL7R+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(1)] <- "CCR7high LEFFhigh SELLhigh"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(2)] <- "NKG7low GZMK+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(3)] <- "FOXP3+ CXCL13+ PDCD1+"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(4)] <- "CD3- NKG7high GNLYhigh NK cells"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(5)] <- "CCR7med LEFFmed SELLmed"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(6)] <- "CCR7low/- LEFFlow SELL-"
# sce_sub$tc_celltype[sce_sub$tc_celltype %in% c(7)] <- "NKG7+ GNLY+/- GZMKhigh/low"
clusters_lookup[[celltype_name_pre]] <- data.frame(cell_id = colnames(sce_sub),
cluster = sce_sub[["tc_celltype"]])tmpfilename <- paste0("syn_v",analysis_version,"_sce_",celltype_name_pre,dplyr::if_else(remove_low_quality_samples, "_invivo",""),".rds")
saveRDS(sce_sub, file = here::here("output",tmpfilename))colData(sce_main)$minor_celltype <- colData(sce_main)$main_celltype
names(clusters_lookup)[1] "sf" "mp" "ec" "tc"for(sub_name in names(clusters_lookup)){
celllabelmatch <- match(clusters_lookup[[sub_name]]$cell_id,
colnames(sce_main))
print(table(is.na(celllabelmatch)))
celllabelmatch <- celllabelmatch[!is.na(celllabelmatch)]
colData(sce_main)$minor_celltype[celllabelmatch] <- clusters_lookup[[sub_name]]$cluster
}
FALSE
30432
FALSE
35659
FALSE
9378
FALSE
23169 dim(sce_main)[1] 17057 102758sce_main <- sce_main[,!is.na(sce_main$minor_celltype)]
dim(sce_main)[1] 17057 102758saveRDS(sce_main, file =here::here("output",paste0("syn_v",analysis_version,"_sce_hvg_cms_doublet_subcluster_invivo.rds")))
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04 LTS
Matrix products: default
BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.8.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] gdtools_0.2.3 tidySingleCellExperiment_1.0.0
[3] SingleCellExperiment_1.12.0 SummarizedExperiment_1.20.0
[5] Biobase_2.50.0 GenomicRanges_1.42.0
[7] GenomeInfoDb_1.26.7 IRanges_2.24.1
[9] S4Vectors_0.28.1 BiocGenerics_0.36.1
[11] MatrixGenerics_1.2.1 matrixStats_0.58.0
[13] magrittr_2.0.1 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] bitops_1.0-6 fs_1.5.0
[3] httr_1.4.2 rprojroot_2.0.2
[5] tools_4.0.3 R6_2.5.0
[7] irlba_2.3.3 vipor_0.4.5
[9] DBI_1.1.1 lazyeval_0.2.2
[11] colorspace_2.0-0 tidyselect_1.1.0
[13] gridExtra_2.3 compiler_4.0.3
[15] git2r_0.28.0 cli_2.3.0
[17] BiocNeighbors_1.8.2 DelayedArray_0.16.3
[19] plotly_4.9.3 labeling_0.4.2
[21] scales_1.1.1 systemfonts_1.0.1
[23] stringr_1.4.0 digest_0.6.27
[25] rmarkdown_2.6 svglite_1.2.3.2
[27] XVector_0.30.0 RhpcBLASctl_0.20-137
[29] scater_1.18.6 pkgconfig_2.0.3
[31] htmltools_0.5.1.1 sparseMatrixStats_1.2.1
[33] highr_0.8 htmlwidgets_1.5.3
[35] rlang_0.4.10 DelayedMatrixStats_1.12.3
[37] generics_0.1.0 farver_2.0.3
[39] jsonlite_1.7.2 BiocParallel_1.24.1
[41] dplyr_1.0.4 RCurl_1.98-1.2
[43] BiocSingular_1.6.0 GenomeInfoDbData_1.2.4
[45] scuttle_1.0.4 Matrix_1.3-2
[47] Rcpp_1.0.6 ggbeeswarm_0.6.0
[49] munsell_0.5.0 fansi_0.4.2
[51] viridis_0.5.1 lifecycle_1.0.0
[53] stringi_1.5.3 whisker_0.4
[55] yaml_2.2.1 zlibbioc_1.36.0
[57] grid_4.0.3 promises_1.2.0.1
[59] crayon_1.4.1 lattice_0.20-41
[61] cowplot_1.1.1 beachmat_2.6.4
[63] knitr_1.31 pillar_1.4.7
[65] glue_1.4.2 evaluate_0.14
[67] data.table_1.13.6 vctrs_0.3.6
[69] httpuv_1.5.5 gtable_0.3.0
[71] purrr_0.3.4 tidyr_1.1.2
[73] assertthat_0.2.1 ggplot2_3.3.3
[75] xfun_0.21 rsvd_1.0.3
[77] later_1.1.0.1 viridisLite_0.3.0
[79] tibble_3.0.6 beeswarm_0.2.3
[81] ellipsis_0.3.1 here_1.0.1